Aktualności
Zdrowie
Alergie
Anatomia
Badania
Badania w ciąży
Biegunka
Borelioza
Choroby genetyczne
Choroby kobiece
Choroby męskie
Choroby oczu i zaburzenia wzroku
Choroby od A do Z
Choroby pasożytnicze
Choroby zakaźne
COVID-19
Cukrzyca
Dermatologia
Gastrologia
Geriatria
Grypa i przeziębienie
Hormony
Kardiologia
Kręgosłup, kości, stawy
Laryngologia
Leki
Neurologia
Medycyna alternatywna
Niezbędnik pacjenta
Objawy
Onkologia
Opryszczka
Ospa
Otyłość
Patologia ciąży
Psychiatria
Testy
Stomatologia i ortodoncja
Układ oddechowy
Układ odpornościowy
Urologia i Nefrologia
Urazy, wypadki
WZW
Inne
Seks
Sztuka kochania
Antykoncepcja
Problemy z seksem
Choroby intymne
Ciąża i dziecko
Leczenie niepłodności
Przed ciążą
Kalendarz ciąży
Poród
Po porodzie
Przebieg ciąży
Zdrowie dziecka
Psychologia
Zdrowie psychiczne
Rozwój osobisty
Emocje
Relacje
Historie pacjentów
Kalkulatory
Kalkulator BMI
Kalkulator ciąży
Kalkulator dni płodnych
Kalkulator kalorii
Kalkulator idealnej wagi
Kalkulator miesiączkowy
Wirtualny alkomat
Uroda
Włosy
Twarz
Ciało
Dłonie i paznokcie
Stopy
Makijaż
Kosmetyki
Zabiegi kosmetyczne
Fit
Ćwiczenia
Diety i żywienie
Rekreacja
Treningi
Premium
Magazyn
#OnkoHero
Gadaj Zdrów
Zdrowo Odpytani
#PodKontrolą
#MEDetektyw
#ŻyjęZ
Głowa do góry
Temat Miesiąca
Newsletter
Uchroni przed rakiem wątroby? "Kosztuje kilkanaście złotych"
"Obudziłam się rano i nie mogłam unieść ręki". Pytamy ortopedę o przyczynę
Popularne zioło na kleszcze jest toksyczne. Halucynacje i zaburzenia pracy serca
Strupki, plamki, krosty. Dermatolożka ostrzega przed mniej znanym rakiem skóry
Aktualności
60 zł wstydu, czyli stomatologia po polsku. Najwyższa Izba Kontroli ujawnia
Przyszła na świat z „rogiem jednorożca”. To przypadek raz na 50 tys. urodzeń
Miliony Polaków przygotowują tak posiłki. Naukowcy alarmują: to szkodliwe dla zdrowia
Cichy zabójca kobiet wciąż ma się w Polsce dobrze. Badanie CA-125 może uratować życie
Zgłosił się na SOR, ale lekarka odesłała go do domu. 30-latek zmarł tuż obok szpital…
Targi Książki i Mediów VIVELO 2024 już w maju na PGE Narodowym! Jakie atrakcje będą …
Wzmacnia odporność, wspiera serce i chroni przed rakiem. Ta odmiana cebuli to skarb
Zamiast białego, sięgnij po czarny. Obniży ciśnienie i wspomoże tarczycę
Genialny sposób na kleszcze. Kosztuje 15 zł, spacerowicze sobie polecają
Ani woda, ani piwo. Naukowcy wskazali, jaki napój najlepiej nawadnia
Christina Applegate odsłania kulisy swojej choroby. „Noszę pieluchy”
Skąd się wzięła żółta woda nad Bałtykiem? Wiemy, czy jest groźna dla zdrowia
Więcej z działu Aktualności
TEMAT MIESIĄCA: MAŁE A GROŹNE. KOMARY, MESZKI I KLESZCZE
Co cię ugryzło? Jak rozpoznać ślady po ukąszeniach owadów [ZDJĘCIA]
Mity na temat kleszczy. Obalamy 14 najpopularniejszych
Ugryzienia meszek - jak je leczyć? Sprawdzone sposoby na ukąszenia meszek
Co przyciąga komary? 9 rzeczy, których musisz unikać!
O tym się mówi
Wycofanie jednej z szczepionek przeciw COVID-19. Podano główny powód
Zażywał fentanyl na dyżurze. Lekarz podpięty pod kroplówkę przyjmował pacjentów na S…
Tak objawia się FLiRT. 13 symptomów nowego podwariantu COVID-19
Jednorazowa wypłata 7000 złotych dla każdego. Skierowano już interpelację
Cykl #żyjęz
Więcej wideo
podcasty mediateka
playlisty mediateka
eska_mediateka
eska2_mediateka
eskarock_mediateka
vibefm_mediateka
vox_mediateka
Nasi Eksperci
Rafał Janiszewski
Właściciel Kancelarii Doradczej świadczącej usługi z zakresu organizacji ochrony zdrowia na rzecz podmiotów leczniczych
Lek. Bartosz Pawlikowski
Specjalista dermatolog, Klinika Pawlikowski, Łódź
Dr hab. n. med. Piotr Rzymski
Biolog medyczny i środowiskowy
Zdrowie
Strupki, plamki, krosty. Dermatolożka ostrzega przed mniej znanym rakiem skóry
Seniorzy mogą liczyć na dodatkowe pieniądze. Wystarczy, że spełnią jeden warunek
43-letniego Roberta Pirosza może ocalić cud i rodacy
To już ostatni moment, by zrobić 12 badań na NFZ za darmo. Sprawdź, kto może skorzys…
Więcej z działu Zdrowie
Choroby zakaźne
Zabójczy wirus przerwał jej karierę. Jak sobie dziś radzi wicemistrzyni olimpijska A…
Już mamy 3 razy więcej zachorowań niż wcześniej przez rok. Powikłania mogą być tragi…
Powikłania po półpaścu mogą towarzyszyć ci do końca życia. Tak się ochronisz
150 milionów infekcji, prawie 1,7 miliona zgonów. Lekooporne grzyby w natarciu
Tajemniczy wirus z TikToka dziesiątkuje Amerykanów? Lekarka: poczekajmy na oficjalne komunikaty
Jak nie zarazić się grypą od domowników? 7 sposobów na bezpieczną zimę
Paciorkowiec - grupy, objawy zakażenia, wywoływane choroby i leczenie
Więcej z działu Choroby zakaźne
Gastrologia
W majówkę sięgasz po smażone mięso i słodkie napoje? Dla ważnego narządu do katastro…
Stres, antybiotyki, zła dieta. Co niszczy florę jelitową i jak ją odbudować?
Stan zapalny jelit może doprowadzić do depresji. Na te objawy koniecznie zwróć uwagę
Dwa niedawno poznane objawy raka trzustki. Pojawiają się nawet rok przed innymi
Żółta kupa nie musi zaraz oznaczać choroby. Ale trzeba to sprawdzić
Krew w kale - przyczyny, objawy i diagnostyka. Co oznacza krew w stolcu?
"Na kolonoskopię kierują, a na gastroskopię nie chcą". Lekarka wyjaśnia dlaczego
Więcej z działu Gastrologia
Leki
"To zagrozi zdrowiu pacjentów". Kolejne zamieszanie w aptekach, lekarze alarmują
Wyłudzili miliony na lekach recepturowych. Zamieszani farmaceuci i lekarze
Wkrótce może zabraknąć ważnych leków dla chorych na raka i padaczkę. "Brak dobrych zamienników"
Epidemia fentanylu w Polsce dopiero się zaczyna. „Uzależnia bardzo szybko”
Uważaj na paracetamol. Może szkodzić sercu nawet w małych dawkach
Nawet 500 tys. zł kary. Rzecznik Praw Pacjenta o kontrowersyjnym leczeniu boreliozy i raka
Popularny lek działa "odchudzająco"? Wyniki są obiecujące
Więcej z działu Leki
Nie przegap
Wszystko o bliznach. Wiesz, jak o nie zadbać?
Materiał sponsorowany
Choroba milionów kobiet. Sprawdź, czy dotyczy i Ciebie
Tekst sponsorowany
Dieta wzmacniająca włosy i paznokcie: o jakie witaminy i minerały musisz zadbać?
Jak dbać o zdrowie, gdy brakuje czasu i siły?
Materiał sponsorowany
Diety i żywienie
To może być najzdrowsza roślina świata. Łagodzi stany zapalne i wspiera mózg
Wyniszcza cerę i układ krążenia, szkodzi trzustce. Polak spożywa 40 kg tej "trucizny"
Wzmacnia odporność, wspiera serce i chroni przed rakiem. Ta odmiana cebuli to skarb
Zamiast białego, sięgnij po czarny. Obniży ciśnienie i wspomoże tarczycę
Przebadali ulubioną przyprawę Polaków. “Góra pestycydów”
Mówią o niej “wyspa długowieczności”. 100-latkowie jedzą 5 tradycyjnych potraw
Ma 64 lata, a wygląda o 20 młodziej. Grażyna Torbicka zdradziła swój sekret
Więcej z działu Diety i żywienie
Kalkulatory
Kalkulator BMI - jak obliczyć wskaźnik BMI?
Kalkulator kalorii - oblicz dzienne zapotrzebowanie kaloryczne
Kalkulator dni płodnych. Jak je obliczyć?
Więcej z działu Kalkulatory
Uroda
Skuteczna pielęgnacja 50+. Na efekty nie trzeba długo czekać
Miały być ratunkiem dla tłustych włosów. Na te kosmetyki uważaj, bo pożałujesz
Od kiedy stosuję te maski, wypadające włosy to nie problem. Wystarczy kilka składnik…
Kiedy skóra potrzebuje nawilżenia, sięgam do kuchennej szafki. Maseczka z płatków dz…
Śpisz w ten sposób? To najprostsza droga do zmarszczek sennych
Działa jak botoks, wystarczy 15 minut. Nie wydasz ani złotówki
Te kosmetyki lubią niskie temperatury. 6 preparatów, które lepiej trzymać w lodówce
Ten wyciąg zdziała cuda na opuchnięte nogi. Naturalne sposoby przegonią zmęczenie
Sprawdzony sposób na truskawkowe nogi. Ten produkt jest w każdej łazience
Najlepszy sposób na oparzenia słoneczne. Babcia zrywała pędy i smarowała nam ramiona
Więcej z działu Uroda
Ciąża i dziecko
9 pytań o gorączkę u dziecka. Znasz wszystkie odpowiedzi?
Zespół Sandifera - co to? Jakie są jego objawy i sposoby leczenia?
Katar u niemowlaka to wyzwanie. Mamy 10 sposobów na zatkany nosek
Alergię pokarmową ma nawet co 10. niemowlę. Jak ją rozpoznać?
Zdrowy sen noworodka kluczem do jego prawidłowego rozwoju
Depresja poporodowa ojców również istnieje. I często jest lekceważona
Lęk separacyjny to problem nie tylko dzieci. Jak sobie radzić?
Więcej z działu Ciąża i dziecko
Seks
Rozporządzenie jest, a farmaceuci mają wątpliwości. Co z pigułką "dzień po"
5 najlepszych pozycji, jeśli penis wydaje się za mały. Nie zawiedziesz się
Spontaniczny czy zaplanowany? Naukowcy już wiedzą, który seks jest lepszy dla związku
Seks analny - chcesz, ale się boisz? Wystarczy wiedzieć te 3 rzeczy
Spadek libido? 5 wskazówek pomoże odzyskać ochotę na seks
Seks oralny, higiena i HPV. Zadbaj o zdrowie penisa
Grubi nie zasługują na seks? Zdradzamy, z czym mierzą się chorzy na otyłość
Więcej z działu Seks
Psychologia
W dzieciństwie „zaczarowano” cię w księżniczkę czy w ogra? Z traumami dzieciństwa mo…
Te 8 rodzajów inteligencji decyduje o sukcesie w życiu. Naukowcy nie do końca zgodni
W piątek nieprzytomni, w poniedziałek nowo narodzeni. Wysokofunkcjonujący alkoholicy dobrze się maskują
Wyniki badań nad śmiechem zaskakują. "Nawet wymuszony ma pozytywny wpływ"
Jaki masz poziom IQ? Sprawdź swoją inteligencję w naszym teście IQ online!
Zapomnieliśmy, że patrzenie prosto w oczy ma wielką moc. "Mąci nam w głowach"
Od 25 lat bada mózg. Naukowiec zdradza 4 rzeczy, które niszczą pamięć
Więcej z działu Psychologia
Badania
Co cię ugryzło? Jak rozpoznać ślady po ukąszeniach owadów [ZDJĘCIA]
Trudności z zajściem w ciążę, problemy ze wzwodem. O tym hormonie mało kto pamięta
Normy cholesterolu w Polsce "są przesadzone"? Nie daj się zwieść, bo zapłacisz zdrowiem
Częsta przyczyna anemii u kobiet. Prosty test da odpowiedź, czy to twój problem
Kręgosłup, a może nerki? Sprawdź, co naprawdę łupie cię w krzyżu. Pomyłki częstsze niż myślisz
Kosztują grosze lub nic, a mogą uratować życie. 6 ważnych badań
Badanie krwi zdradzi, czy grozi ci rak tarczycy. Taki wynik powinien wzbudzić czujność
Więcej z działu Badania
Fit
Dieta 6:1 przyda się po majówce? Nowy trend pod lupą dietetyka
Spalisz tłuszcz, wesprzesz serce i skołatane nerwy. Uważaj jednak, jeśli masz proble…
Odkryto „wyłącznik” brunatnej tkanki tłuszczowej. Rozpracowanie go pomoże w walce z …
Nogi Beaty Kozidrak powalają. Ograniczyła w diecie jeden składnik. "Ale jem wszystko"
Efekty jak przy ozempiku, ale bez leków. Naukowcy ustalili, co i jak jeść, żeby chud…
"Najlepszy detoks w proszku". Jak naprawdę działa zielony, modny suplement
Schudła 44 kg w 15 miesięcy. Alessia zdradza swój sekret na szczupłą sylwetkę
Ma regenerować organizm, świetnie wpływać na jelita. Koniec z bulgotami, biegunkami …
Królowa Elżbieta dożyła niemal 100 lat. Jej sekret tkwił w tych rytuałach
Odchudzasz się, a waga się zatrzymała i nie spada? Ekspert wie, dlaczego tak się dzi…
Magdalena Lamparska pozbyła się 10 kilogramów. Zdradziła, jak to osiągnęła
Ma 60 lat, a ciało 38-latka. Wyjawił swoje patenty na zachowanie tak młodej formy
Więcej z działu Fit
Patronaty
„SM - i co z tego…?”. Życie nie kończy się po diagnozie
To uważność kobiety decyduje o zdrowiu rodziny. Często nie starcza jej siły dla sieb…
Poznaj swój mózg. Powstał poradnik dla pacjentów
Więcej z działu Patronaty
Współpracują z nami:
Polskie Towarzystwo Okulistyczne
Fizjoterapia Polska
Warsztaty Kardiologii Interwencyjnej
Fundacja Odwaga
Fundacja SMA
Zaszczep się wiedzą
Polskie Towarzystwo Do Badań Nad Rakiem Piersi
Bieg po nowe życie
Fundacja Zdrowie Jest Najważniejsze
Kampania Stawka to życie. Zastawka to Życie
Ifmsa-Poland
Koalicja na rzecz walki z łuszczycą
Europa Colon Polska
Mazowiecki Oddział Okręgowy Polskiego Czerwonego Krzyża
Konferencja Powstrzymać smog. Wyzwania i Rozwiązania dla Polski
Kampania Kardiologia bez kolejki
email















![Co cię ugryzło? Jak rozpoznać ślady po ukąszeniach owadów [ZDJĘCIA]](https://cdn.galleries.smcloud.net/t/galleries/gf-hqBv-jvWR-tGQB_co-cie-ugryzlo-jak-rozpoznac-slady-po-ukaszeniach-owadow-zdjecia-1008x442.jpg)
















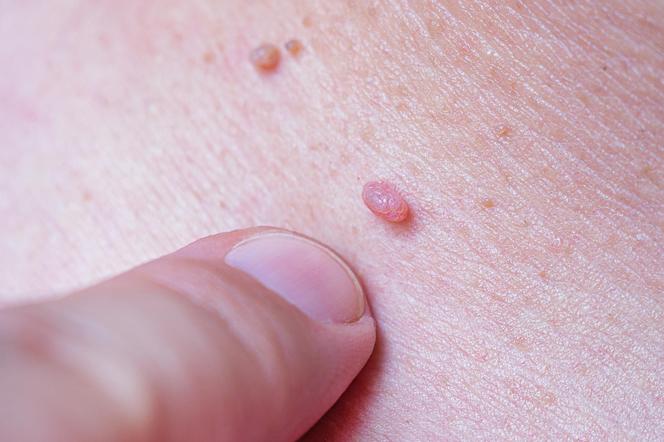











































![Co cię ugryzło? Jak rozpoznać ślady po ukąszeniach owadów [ZDJĘCIA]](https://cdn.galleries.smcloud.net/t/galleries/gf-uFE7-Cc8o-iwRF_co-cie-ugryzlo-jak-rozpoznac-slady-po-ukaszeniach-owadow-zdjecia-320x213.jpg)